Main
TnpBs are a family of RNA-guided endonuclease proteins encoded within IS200/IS605 and IS607 transposons and are thought to be the evolutionary ancestors of type V CRISPR–Cas enzymes1,2,3,4,5. TnpB binds to the right-end element RNA (reRNA), which has a noncoding 5′ scaffold and a variable 3′ region, to guide ribonucleoprotein (RNP) cleavage at a cDNA sequence proximal to a transposon-associated motif (TAM)2,3. As a putative evolutionary predecessor to the CRISPR–Cas12 enzymes, TnpB retains core domains shared across this CRISPR–Cas protein family4,5,6,7. Understanding the relationship between the protein sequence and activity of TnpB can provide both fundamental knowledge and serve as a basis for engineering improved or altered RNA-guided endonucleases.
While highly active variants of CRISPR–Cas enzymes have been identified through protein engineering and rational design8, these approaches often explore a limited sequence space. The conformational changes TnpB undergoes during the dynamic coordination of nucleic acid binding, catalytic center activation and DNA cleavage6,7 make it challenging to predict the effects of mutations. Deep mutational scanning (DMS) approaches typically assess every individual amino acid mutation using high-throughput assays of protein function9,10,11. While DMS effectively provides comprehensive maps of protein function, it is often practically limited by protein size. TnpB is uniquely well suited for this approach because of its compact amino acid length and RNA scaffold.
We conducted DMS over the entire ISDra2 TnpB RNP, one of the first experimentally characterized TnpB orthologs2. Using a positive selection assay for DNA cleavage, we identified a broad spectrum of enhancing, neutral and deleterious mutations within the TnpB protein and its reRNA. These data elucidate dynamic regions involved in DNA binding and cleavage, including a mutational hotspot within the reRNA secondary structure where mutations increase DNA cleavage activity. We found that 20% of single-amino-acid substitutions, many of which are not frequently observed in nature, increase activity relative to the wild-type (WT) TnpB protein. This suggests that native ISDra2 TnpB activity may be subject to negative selection, possibly because of its role as a transposon-associated homing endonuclease2,12. Furthermore, we identified combinations of activating mutations that increase TnpB-mediated genome-editing activity in both human cells and plants.
Results
Selection for TnpB-mediated DNA cleavage in yeast
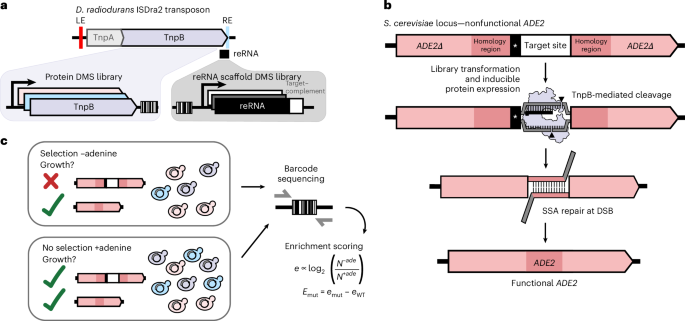
In their native genomic context, the TnpB reRNA and protein-coding sequences overlap with each other and with insertion sequence elements essential for transposition, imposing unknown sequence–function constraints2,3. To interrogate the effects of mutations in both reRNA and TnpB protein without the native sequence constraints of transposition, we encoded the reRNA and codon-optimized ISDra2 TnpB protein under the control of separate regulatory elements (Fig. 1a). To assess on-target cleavage activity, we adapted an in vivo selection previously used to enhance CRISPR–Cas9 activity, which uses yeast (Saccharomyces cerevisiae) strains with a genomic ade2− reporter cassette13 (Fig. 1b). On-target cleavage of the reporter cassette initiates ADE2 repair, enabling cells to grow on medium lacking adenine. Thus, reporter strain growth in the presence and absence of adenine can be used as a readout for target site cleavage (Fig. 1c). We first demonstrated that this assay can quantitatively measure endonuclease activity beyond that of CRISPR–Cas9, including WT ISDra2 TnpB and CRISPR–Cas12 endonucleases13 (Extended Data Fig. 1a).

a, TnpB protein and reRNA from the D. radiodurans ISDra2 transposon were placed under the control of separate regulatory elements. DMS libraries were constructed for both molecules and assayed separately. b, Schematic of the yeast-based cleavage assay used for individual variant testing and high-throughput library experiments. On-target double-stranded breaks in the reporter cassette enable repair of the ADE2 locus by single-stranded annealing at duplicate homology regions. ADE2 repair rescues colony growth in selective, adenine-deficient medium. c, Representation of the library selection. Plasmid DNA from yeast grown in selective (−adenine) and nonselective (+adenine) conditions was extracted. Barcodes from plasmids were sequenced and the log ratio of barcode abundance in selective over nonselective conditions was used to assess variant enrichment. Enrichment of variants was normalized to the WT TnpB RNP enrichment.
We constructed independent pooled plasmid libraries of reRNA and protein variants (Fig. 1a). The reRNA and protein DMS libraries were barcoded such that each variant was associated with ~30 unique barcodes, providing statistical replicates. The libraries were transformed into yeast reporter strains with their WT reRNA or protein counterparts and barcode abundance in selective and nonselective media was quantified at multiple time points across two biological replicates. Relative variant enrichment was calculated as the log ratio of variant abundance in selective and nonselective conditions and all enrichments were normalized to WT controls (Fig. 1c).
Profiling the mutational landscape of the TnpB reRNA
The reRNA accounts for nearly half of the molecular weight of the ISDra2 TnpB RNP complex6,7. Previously, a truncation within the reRNA stem 2 region, termed Trim2, was shown to maintain or increase the ISDra2 TnpB genome-editing activity7,14 (Extended Data Fig. 1b). Given the complex secondary and tertiary structures of the TnpB RNA scaffold, we hypothesized that comprehensive mutagenesis of the reRNA scaffold could reveal insights into evolutionary constraints on the reRNA sequence and RNP endonuclease activity.
The 116-nt reRNA scaffold is necessary and sufficient for TnpB-mediated cleavage7 and was chosen as the starting sequence (termed WT reRNA) for DMS (Extended Data Fig. 1c). To investigate the mutational tolerance of the reRNA, our DMS library included all single-nucleotide substitutions, as well as both single-nucleotide and double-nucleotide deletions. We also included variants with the disordered regions of stem 1 and stem 2 replaced with thermodynamically stable tetraloops15,16, the reported Trim2 variant and variants with reported inactivating truncations within the triplex and pseudoknot serving as negative controls14, for a total of 576 assayed mutants (Fig. 2a,b).

a, Schematic of the ISDra2 reRNA based on the RNP cryo-EM structure, adapted from Sasnauskas et al.6. Dashed boxes indicate truncations replaced with stable tetraloops. Circles around bases are colored by maximum log2-transformed enrichment scores for a substitution at each position, using the same color scale as in c. Outer gray-circled bases indicate regions not modeled in the ternary structure, labeled here as disordered. b,The log2-transformed enrichment of reRNA variants across two experimental replicates. Data are normalized such that WT TnpB is at 0. c, Heat map of enrichment scores for reRNA variants with single-nucleotide substitutions or single-nucleotide and double-nucleotide deletions. WT positions are colored in white, while gray boxes denote no available data (ND). d, Structure of TnpB ternary complex (PDB 8EXA), with reRNA colored by positional maximum log2-transformed enrichment scores. e, Experimental workflow of EGFP KO assay in HEK293T EGFP+ cells. EGFP− cells were assessed by flow cytometry to compare editing activities of TnpB variants, as described in the Methods. f, EGFP KO assay in HEK293T EGFP+ cells for highly enriched reRNA mutants identified with bold black outlines in b. Colors match the legend in b. The fold change of EGFP− percentage population for each variant compared to WT TnpB is shown. Data are presented as the mean ± s.e.m. from biological transfection replicates (n = 3). Asterisks indicate a statistically significant increase in indel frequencies compared to WT ISDra2 reRNA as calculated using a two-sided unpaired Student’s t-test.*P ≤ 0.05, **P ≤ 0.01 and ***P ≤ 0.001.
Upon mapping the reRNA mutational landscape, we found that inactivating truncations and deletions within the pseudoknot were depleted, as expected, under selective conditions (Fig. 2b). Stable tetraloop replacements in stem 2 were more highly enriched compared to those in stem 1 and Trim2 was one of the most highly enriched variants. We observed substitutions and deletions that were more enriched than both WT and the Trim2 variant. These activating mutations were concentrated around unpaired nucleotides rA−40–rU−43 within stem 2 (Fig. 2a,c). We refer to this region (rA−37–rU−44; rG−75–rG−79) as the ‘hinge’ region of the reRNA, which appears to create a sharp bend in stem 2 preceding the disordered distal end6 (Fig. 2d). We also tested reRNA variants in HEK293T cells using an enhanced green fluorescent protein knockout (EGFP KO) assay17. When targeted to an EGFP transgene, reRNA variants with nucleotide deletions in the hinge region resulted in the greatest increase in EGFP KO compared to the WT reRNA, as assessed with flow cytometry (Fig. 2e,f). By contrast, reRNA variants with a truncation in stem 2, including Trim2, showed an unexpected decrease in EGFP KO efficiency compared to the WT reRNA, despite being highly enriched in the yeast selection (Fig. 2b,f).
Stem 2 was proposed by Sasnauskas et al. to act as a regulatory switch that controls the transition of the TnpB RNP into a cleavage-competent conformation upon DNA binding and heteroduplex formation6. This activation is driven by a conformational change in stem 2, where the formation of the RNA–DNA heteroduplex displaces the distal end of stem 2, leading to the release and activation of the RuvC domain (Supplementary Data Fig. 1). We speculate that activating hinge mutations enhance TnpB activity by increasing the flexibility of the distal end of stem 2, making it more prone to displacement. This could facilitate the release and activation of the RuvC domain. This aligns with the previous report that truncation of stem 2 also leads to dysregulated collateral single-stranded DNA (ssDNA) cleavage, independent of target DNA binding6. Variability in editing levels that we observe with stem 2 truncation variants may also support the importance of stem 2 in regulating reRNA-mediated TnpB activity (Fig. 2f). However, the precise mechanism underlying increased activity of reRNA variants remains unclear and requires further biochemical and structural investigation.
DMS of TnpB protein
We next mapped the fitness landscape of the ISDra2 TnpB protein with a library spanning all possible single-amino-acid substitutions and stop codons, alongside catalytically inactive (dead) and WT protein controls (Fig. 3). We collected data on 93% (7,611 of 8,140) of all possible substitutions across two biological replicates, which were highly reproducible with a Pearson correlation of 0.81 (Fig. 4a). We found that 3.7% of these substitutions were enriched at least twofold compared to WT TnpB (Figs. 3 and 4a).

Heat map showing log2-transformed enrichment scores of all single-amino-acid changes. TnpB secondary structures and domains are annotated above the heat map. Outlined white boxes represent WT residues. Gray boxes denote positions with no available data. Boxes with a slash and an asterisk at the x axis represent a mutation that was included in the combinatorial variant library. Black triangles represent active-site catalytic residues.

a, The log2-transformed enrichment of single-amino-acid variants across two experimental replicates. Data are normalized such that WT TnpB is at zero. A total of 33 mutations were selected from the combinatorial library. Purple points correspond to alanine substitutions of the TAM-interacting residues (Y52A, K76A, Q80A, T123A, S56A, F77A and N124A) shown to abrogate or reduce TnpB activity7. b, Close-up view of residues N4, P282, E302 and I304 in red in the ISDra2 TnpB ternary structure (PDB 8EXA). RuvC catalytic residues are colored and labeled in dark blue. Site-wise amino acid enrichment and MSA conservation are displayed on radar plots below. N4 MSA data are not shown because of low sequence conservation at the N terminus. c, Maximum enrichment per residue mapped onto the surface of ISDra2 TnpB ternary structure, shown as a cross-section (left), allowing for visibility of the heteroduplex (PDB 8EXA). The reRNA is shown in gray and DNA is shown in light yellow. d, Activity of the highly enriched single-amino-acid mutants was assessed with the EGFP KO assay in HEK293T EGFP+ cells. Fold change of EGFP− cells (percentage of population) for each variant relative to WT TnpB is shown. Data are presented as the mean ± s.e.m. from biological replicates (n = 3). Stars indicate a statistically significant increase in indel frequencies compared to WT ISDra2 TnpB as calculated using a two-sided unpaired Student’s t-test. *P ≤ 0.05, **P ≤ 0.01 and ***P ≤ 0.001.
Enrichment density varied across domains, with many mutations enriched in the RuvC and WED domains, most ZnF mutations depleted and mutations in the unstructured C-terminal tail largely neutral (Fig. 3 and Extended Data Fig. 2a). Stop codons causing truncations were depleted except for those at the C terminus following residue 376, consistent with previous in vitro data showing that the C-terminal tail is dispensable for target cleavage7 (Extended Data Fig. 2b). Additionally, we observed depletion of alanine substitutions at residues important for recognizing the TAM, a 5′-TTGAT-3′ sequence that is essential for cleavage at the adjacent target sequence2,3,7 (Fig. 4a).
Positively charged amino acids were enriched over negatively charged residues within the vicinity of nucleic acids, particularly within the central channel, where the TAM-proximal end of the heteroduplex is accommodated6 (Extended Data Fig. 3). This is consistent with previous findings in CRISPR–Cas12 enzymes, where mutations introducing positively charged amino acids near the guide RNA–DNA heteroduplex, close to the protospacer-adjacent motif (PAM), have been shown to increase activity and affect specificity18,19.
Similarly, specific WED-domain residues, N4 and L172, which stabilize the first TAM-proximal base pair of the heteroduplex6, were enriched for aromatic and small functional groups, respectively (Fig. 4b). We reason that the initial reRNA–target duplex formation may be enhanced by small hydrophobic and nucleophilic amino acids at position 172, and by π-stacking interactions between aromatic residues at N4 and the first heteroduplex nucleobases. The introduction of aromatic amino acids near the first base pair of the RNA–DNA heteroduplex has also been associated with increased activity in AsCas12f (refs. 20,21), providing evidence for a shared activation mechanism within an interaction conserved across TnpB and CRISPR–Cas12f endonucleases.
Within the positively charged central channel, E302 was highly enriched for substitutions to any amino acid that was not negatively charged (Fig. 4b). Positioned near the heteroduplex backbone, E302 might lead to electrostatic repulsion with the target strand (TS) phosphate backbone, potentially reducing RNP activity. We also identified other mutational hotspots, such as I304, where substitutions were enriched for residues with a range of physicochemical properties.
Hydrophobic amino acids were enriched at position P282, which lies at the boundary of the lid subdomain that blocks the RuvC active site from accessing the TS6 (Figs. 3 and 4b). The lid subdomain forms non-sequence-specific contacts with the heteroduplex minor groove, which may aid in sensing heteroduplex formation before RuvC activation. We hypothesize that substituting the WT proline residue at this position with small, hydrophobic residues could increase the flexibility of the lid subdomain and accelerate the conformational change required to sense heteroduplex formation before TnpB activation.
Overall, 844 mutations from the DMS dataset were enriched over WT TnpB with a P value < 0.05 and were distributed across both the nucleic-acid-binding interface and the protein surface (Fig. 4c). We tested 20 highly enriched mutants in HEK293T cells with the EGFP KO assay. All reduced EGFP expression, with P282I resulting in a nearly fourfold reduction relative to WT TnpB (Fig. 4d).
To explore the generalizability of this protein DMS dataset, we transferred pairs of activating mutations to TnpB orthologs ISYmu1 and ISAba30 (ref. 22). These proteins share notable structural similarity with ISDra2 TnpB (pairwise TM-scores of 0.93 and 0.92 with ISYmu1 TnpB and ISAba30 TnpB, respectively), despite low sequence similarity (59% with ISYmu1 and 48% with ISAba30)23,24. We designed two ISYmu1 TnpB variants (H4Y;V305R and L167G;V305R) and one ISAba30 TnpB variant (L4Y;V272I) by introducing pairs of analogous activating substitutions from ISDra2 TnpB (N4Y/I304R, L172G/I304R and N4Y/P282I, respectively) (Extended Data Fig. 4a). We observed increased colony reversion with all three variants compared to their WT orthologs in the yeast cleavage assay (Extended Data Fig. 4b,c). These results demonstrate that these activation mechanisms are generalizable beyond ISDra2 TnpB, underscoring the utility of our mutational dataset for informing further engineering and characterization of diverse systems.
Combinatorial mutations enhance TnpB activity
To explore increases in TnpB editing activity through mutation combinations, we selected 33 highly enriched single-amino-acid substitutions covering 19 positions across TnpB (Fig. 3). Using nicking mutagenesis, we generated a library of ~5 × 103 variants with an average of ~5 of the 33 possible mutations per variant25,26 (Extended Data Fig. 5a). This combinatorial variant library underwent selection in two reporter yeast strains with different target sequences (Fig. 5a, Extended Data Fig. 5b). We observed the greatest increase in enrichment in variants with 4–5 mutations on average, while depleted variants had more variable mutation numbers (Extended Data Fig. 5c). The expression levels of seven highly active TnpB variants were found to be similar to WT by western blot, consistent with a change in enzymatic activity rather than protein abundance (Extended Data Fig. 6).

a, Volcano plot depicting combinatorial variant average enrichment and statistical significance, after selection. Individual barcodes were used to calculate significance with a two-sided Mann–Whitney U-test. b, Indel frequencies of five combinatorial variants at four genomic loci in HEK293T cells. TnpB variants were delivered by transfection and genomic DNA was sequenced as described in the Methods. c, Indel frequencies of TnpB variants targeting two sites in PDS1 in N. benthamiana. Legend, same as in b. d, Indel frequency of WT ISDra2, TnpB-KYLI and TnpB-VGIRL targeting eight sites in N. benthamiana. Data are plotted as the mean ± s.e.m. from independent biological replicates (n = 3) in b–d. e, Indel frequencies in rice plantlets regenerated from callus tissue stably transformed with TnpB variants targeting three genomic sites. Average indel frequencies for individual transformation events were calculated as described in the Methods. Data are plotted as the mean ± s.e.m. from independent biological replicates: OsHMBPP, n = 6, 7 and 5; OsPDS, n = 14, 6 and 9; OsSLA4, n = 4, 10 and 8 (left to right). f, Indel frequency of TnpB variants targeting two sites in pepper. Data are plotted as the mean ± s.e.m. from biological replicates (n = 4 for all conditions except eTnpBe at CaCHLH-2, where n = 5). TnpB variants were delivered by agroinfiltration and indel frequencies were quantified from tissue as described in the Methods for c–f. Asterisks indicate a statistically significant increase in indel frequencies compared to WT ISDra2 TnpB as calculated using a two-sided unpaired Student’s t-test (b–d,f) or two-sided Welch’s t-test (e). *P ≤ 0.05, **P ≤ 0.01 and ***P ≤ 0.001.
To assess the genome-editing activity of TnpB combinatorial variants, we selected five highly active variants (eTnpBa–eTnpBe) for testing at five genomic loci in human cells (Fig. 5b and Extended Data Fig. 7a). HEK293T cells were transfected with plasmids encoding each TnpB protein variant targeting endogenous loci and indel (insertion and deletion) frequencies were assessed 4 days after transfection. Compared to WT TnpB, all five variants demonstrated higher indel formation frequencies across all target sites, except for eTnpBc (N4Y;R110K;V192L;L222I) at the AGBL1 locus. Variant eTnpBd (R110K;P282V;E302Q) achieved the highest overall indel frequencies across multiple loci (23–42%), surpassing both WT ISDra2 and ISYmu1 TnpB (11–29% and 6–30%, respectively) (Fig. 5b and Extended Data Fig. 7a).
To evaluate off-target activity in HEK293T cells, we identified six genomic sites with 4–6 mismatches to the target-complementary reRNA sequence with Cas-OFFinder27. All variants exhibited increased off-target indel frequencies compared to WT ISDra2 and ISYmu1 on at least two sites, with up to 6% off-target indel frequencies observed with eTnpBc at TET1 off-target site 1 (Extended Data Fig. 7b). Generally, lower indel frequencies occurred at off-target sites with more TAM-proximal mismatches preceding the 12th nucleotide, consistent with published data indicating that TAM-distal mismatches are more well-tolerated by WT TnpB6,28. While increases in on-target editing were consistently accompanied by increases in off-target indel frequencies, eTnpBe (L172G;V192L;L222I;P282V;I304R) was associated with the lowest off-target activity (<2% at all sites) of the variants.
We investigated whether genome-editing activity could be further enhanced by combining highly active TnpB protein with reRNA variants (Extended Data Fig. 7c). Using the EGFP KO assay, we tested five TnpB protein mutants paired with one of two reRNA mutants (ΔrU−42 or ΔrC−74–rG−75), each with deletions on the 5’ or 3’ side of the hinge region. However, many of these pairings with the reRNA mutants did not result in substantial additive improvements in editing, compared to pairings with the WT reRNA. Instead, combining the highly active eTnpBd variant with either hinge deletion variant resulted in reduced genome editing (15–24% EGFP− cells) compared to the WT reRNA (44.5% EGFP− cells). One possible explanation is that certain protein–reRNA variant combinations destabilize the TnpB RNP beyond a critical free energy threshold, disrupting RNP assembly or dsDNA targeting29. Further studies are needed to understand how protein and reRNA mutations interact to influence RNP stability and function. Systematic combinatorial testing of reRNA and protein variants may reveal more optimal pairings that enhance RNP stability and activity.
Enhanced TnpB variants for genome editing in plants
Precise genome editing offers major advantages over traditional breeding in identifying and developing novel crop traits but is still limited by efficient delivery of gene-editing components and the low throughput of plant tissue culture30,31. Viral vectors have shown promise in delivering gene-editing reagents to induce heritable germline edits across various species; however, their cargo capacity remains a notable limitation for delivering standard CRISPR–Cas enzymes32,33. TnpB, because of its compact size, is well suited to overcome this barrier. Multiple TnpB orthologs, including ISDra2, have shown potential for genome editing in plants but WT editing efficiencies remain low34,35,36.
To assess their utility for plant genome editing, we tested eTnpBa–eTnpBe in the model dicot Nicotiana benthamiana. TnpB variants and reRNA targeting three sites within NbPDS1 (phytoene desaturase) were delivered into N. benthamiana leaves by agroinfiltration and indel frequencies were assessed within the infiltrated leaf tissue. All five variants exhibited increased editing activity at NbPDS1-1 and NbPDS1-4 sites (Fig. 5c). At both NbPDS1-1 and NbPDS1-4, eTnpBc demonstrated the highest editing efficiencies of (33% and 45%) compared to WT TnpB (<1% and 6%), with all variants demonstrating editing levels between roughly 4–40-fold higher than WT levels. At the NbPDS1-2 site, an increased indel frequency was observed for eTnpBe (8% versus 2% WT TnpB) (Extended Data Fig. 8a). By contrast, we did not observe substantial increases in editing with the reRNA and single-amino-acid substitution variants compared to WT ISDra2 at NbPDS1-1 and NbPDS1-2 (Extended Data Fig. 8b).
On the basis of their activity across multiple genomic sites in HEK293Ts and N. benthamiana, we selected eTnpBc and eTnpBe, hereafter referred to as TnpB-KYLI (R110K;N4Y;V192L;L222I) and TnpB-VGIRL (P282V;L172G;L222I;I304R;V192L), for assessment at eight additional genomic target sites in N. benthamiana (Fig. 5d). Except for TnpB-KYLI at the NbDMR6 site, both TnpB-KYLI and TnpB-VGIRL exhibited increased editing activity compared to WT ISDra2 at all sites, with TnpB-KYLI and TnpB-VGIRL reaching over 50-fold increases in indel frequencies (55% and 49%) compared to WT ISDra2 (<1%) at NbNDR1. At the NbDMR6 site, TnpB-VGIRL exhibited an increased indel frequency (23%) relative to WT ISDra2 (4%). Furthermore, editing levels of TnpB-KYLI and TnpB-VGIRL at off-target sites predicted by Cas-OFFinder27 were comparable to or lower than those observed for the WT TnpB, indicating that the engineered variants maintain target specificity in N. benthamiana (Extended Data Fig. 9a).
We compared the editing efficiencies of TnpB-KYLI and TnpB-VGIRL with WT ISDra2 and ISYmu1 TnpB, as well as other recently engineered small RNA-guided endonucleases, such as AsCas12f-HKRA21 and NovaIscB37, which also offer advantages for viral delivery where cargo size is limited. Among these small RNA-guided endonucleases, TnpB-VGIRL and TnpB-KYLI showed the highest editing levels at all three target sites in N. benthamiana, with TnpB-VGIRL nearly matching the indel frequencies observed with Cas9 at NbWRKY40 (38% and 39% for TnpB-VGIRL and Cas9, respectively) (Extended Data Fig. 9b). In comparison, the editing levels of AsCas12f-HKRA, NovaIscB and ISYmu1 were consistently lower (<9%) or undetectable.
We speculate that the relatively low editing efficiencies of AsCas12f-HKRA and NovaIscB in N. benthamiana may be because of differences in the strategies used to engineer these RNA-guided endonucleases for increased activity. While AsCas12f-HKRA and NovaIscB were optimized using HEK293T cell-based assays or in vitro cleavage assays at or above 37 °C (refs. 21,37), our selections were performed in yeast, which grow at 30 °C, and may have enriched for variants with greater activity at lower temperatures optimal for plant growth (23–28 °C; Methods).
Lastly, we investigated the ability of the variants to edit in the agriculturally important monocot and dicot crop species, rice (Oryza sativa) and pepper (Capsicum annuum). To assess the potential for generating stable transgenic lines, we measured editing in rice leaf tissue regenerated from rice calli stably transformed with Agrobacterium carrying TnpB-KYLI, TnpB-VGIRL and WT TnpB targeting three genomic loci. TnpB-KYLI and TnpB-VGIRL showed higher editing levels than WT TnpB at all three sites, with up to 25.3% and 29.3% indel frequencies observed for TnpB-KYLI and TnpB-VGIRL at OsHMBPP (Fig. 5e). Although rice callus transformation methods are well established and WT ISDra2 TnpB editing has been reported in rice, effective delivery of genome-editing components into pepper remains limited38,39,40. To further demonstrate editing activity of TnpB-KYLI and TnpB-VGIRL in a nonmodel crop, we delivered these variants and reRNA targeting five genomic sites into pepper leaves by Agrobacterium infiltration. TnpB-KYLI and TnpB-VGIRL demonstrated consistently higher editing compared to WT TnpB at all five sites, with up to 10% editing with TnpB-VGIRL at CaAGO2 compared to <1% editing with WT TnpB (Fig. 5f and Extended Data Fig. 9c). Overall, these findings highlight the potential of TnpB-KYLI and TnpB-VGIRL for high-efficiency editing in both crops and further optimization of delivery strategies may enable higher editing levels.
Discussion
In the Deinococcus radiodurans genome, ISDra2 TnpB is encoded alongside the HUH superfamily TnpA transposase, with both relying on overlapping sequences essential for transposition and endonuclease activity2,41. The reRNA stem 1 sequence overlaps with the imperfect hairpin in the transposon right end, which is required for TnpA recognition and excision6,41,42. Notably, in our DMS datasets, stem 1 exhibited increased mutational tolerance, as did the few protein residues within the vicinity of stem 1, supporting the hypothesis that TnpB protein and reRNA have coevolved with TnpA and the transposon43 (Extended Data Figs. 3a and 10).
The selective balance of transposon maintenance, propagation and effects on host fitness may constrain TnpB nuclease activity in its native setting12,44,45. By profiling TnpB-mediated on-target cleavage outside the context of transposition, we identified many activating mutations across the RNP, highlighting the rugged nature of the mutational landscape7,46. The frequency of activating and neutral mutations in TnpB is an outlier compared to standard models of protein evolution and other mutational studies29,47,48,49. This aligns with the hypothesis that TnpB exhibits pervasive evolutionary flexibility, having been exapted for diverse biological processes across multiple clades of life1,43,50,51. Additionally, the prevalence of evolutionarily accessible activating mutations may suggest TnpB endonuclease activity is under negative selective pressure in the transposon context.
This work presents comprehensive sequence–function landscapes for both the protein and RNA scaffold of an RNA-guided endonuclease. Comprehensive reRNA mutagenesis uncovered an unexpected mutational hotspot in stem 2 and offers an alternative approach to iterative reRNA and gRNA engineering through truncations and G:U swaps to optimize gene-editing activity6,21,52. Mutational scanning of the TnpB protein not only reproduced published findings on ISDra2 TnpB point mutants7,46 but also captured additional activating mutations that increased on-target cleavage activity. We further demonstrate that activating mutations can be combined to enhance genome-editing activity in HEK293T cells, N. benthamiana, rice and pepper, and we present TnpB-KYLI and TnpB-VGIRL as highly active variants.
We recently demonstrated that viral delivery of TnpB-KYLI in N. benthamiana drives high somatic editing and yields >50% heritable edits in offspring, markedly outperforming WT ISDra2 TnpB53. These findings establish engineered, compact TnpB variants as a promising solution for highly efficient, transgene-free, heritable genome editing in plants where cargo size has been a major limitation31. Further exploration of alternative delivery methods and editing levels in diverse crop species and broadening of the 5′-TTGAT-3′ TAM recognition motif will increase the utility of TnpB variants for genome-editing applications32.
Overall, these comprehensive mutagenesis libraries provide molecular insights into nucleic acid binding, activation and cleavage by TnpB, mapping both mutational constraints and activating mutations across the RNP. Further biochemical and epistatic studies may help elucidate mechanisms of activating mutations in TnpB and related endonucleases. In addition to laying the groundwork for further engineering, we hope our findings will provide insights into the evolution and function of TnpB within insertion sequences.
Methods
Deep mutational library construction
The TnpB reRNA DMS library was constructed from an oligonucleotide pool from Twist Bioscience and covered the 116-nt reRNA scaffold with flanking primer-binding sites for PCR amplification. The reRNA scaffold library contained ~600 variants, including all nucleotide substitutions, single-nucleotide and double-nucleotide deletions, a set of double mutations in the pseudoknot and stable tetraloop replacements in the disordered reRNA regions. The oligonucleotide library was amplified using KAPA HiFi HotStart ReadyMix with an initial denaturation at 95 °C for 3 min, followed by 16 cycles of 98 °C for 20 s, 64 °C for 15 s and 72 °C for 45 s, with a final extension at 72 °C for 1 min. The amplified library was cloned into an intermediate storage vector with NEBuilder HiFi DNA assembly master mix. The reRNA library was then assembled with a destination vector containing the variable 3′ reRNA sequence by Golden Gate cloning with BsaI-HFv2 and MlyI.
The reRNA plasmid library was digested with KpnI and ApaI and barcoded by Gibson assembly (NEBuilder HiFi DNA assembly master mix) with ssDNA oligonucleotides with internal 15 × N barcodes. The barcoded plasmid assembly was transformed into TransforMax EC100D pir-116 electrocompetent Escherichia coli and bottlenecked such that a larger culture for plasmid purification was inoculated with ~2.4 × 104 transformed cells (~40 barcodes × ~600 variants), estimated from colony-forming units (CFUs) counted from titer plates. Control plasmids containing WT and catalytically dead ISDra2 TnpB were barcoded similarly.
The ISDra2 TnpB protein sequence was codon-optimized for expression in S. cerevisiae and human cells and divided into six segments of 204 bp. For each segment, mutations for all single-amino-acid changes and stop codons were designed and purchased as oligonucleotide pools from Twist Bioscience with flanking primer-binding sites, for a total of 8,116 variants. To account for enrichment of truncations at all stop codons, the nuclear localization sequence tag was positioned at the N terminus of the TnpB protein in the protein libraries, instead of the C terminus, where it was placed in the reRNA library. Mutations were designed using the most common S. cerevisiae codons, except in cases where this would create a restriction site that would interfere with library cloning or plasmid linearization. In these cases, an alternative common codon set was used to introduce the intended mutation. The first methionine was excluded from mutagenesis. The six sublibraries were amplified using KAPA HiFi HotStart ReadyMix with an initial denaturation at 95 °C for 3 min, followed by 18 cycles of 98 °C for 20 s, 64–67 °C for 15 s and 72 °C for 45 s, with a final extension at 72 °C for 1 min. Amplified sublibraries were assembled by Golden Gate cloning with BsaI-HFv2 and six corresponding intermediate cloning vectors containing the flanking WT ISDra2 sequence. Each sublibrary plasmid pool was digested with BsmBI-v2 and the concentration of the digested full-length ISDra2 protein-coding sequence for each sublibrary was measured using a Qubit 4 fluorometer. Each digested sublibrary was mixed at an equimolar ratio and inserted into the destination vector.
Single-stranded 30 × N barcode sequences were cloned into the NotI-HF-digested and XhoI-digested plasmid library with NEBuilder HiFi DNA assembly master mix. Assemblies were transformed into TOP10 electrocompetent E. coli and a larger culture for plasmid purification was inoculated with ~2 × 105 transformed cells (~24 barcodes × ~8,100 variants).
Combinatorial library construction
Two combinatorial libraries with an average of ~3 and ~5 mutations per variant were created using nicking mutagenesis25,26. DNA oligos covering 19 amino acid positions and 33 possible mutations in the TnpB protein were phosphorylated and pooled in an equimolar ratio. For synthesis of the second strand, 5 pmol and 50 pmol of the phosphorylated oligo pool was initially added with 0.38 fmol of the ssDNA template plasmid and 4.3 pmol and 43 pmol of the phosphorylated oligo pool was spiked in three times following five cycles of amplification, to generate the libraries with lower and higher mutation frequency, respectively.
RRY(N × 25)RY and YYR(N × 25)YR barcodes were cloned into the plasmid libraries with lower and higher mutation frequency, respectively, by assembly with ssDNA oligos, as described above. The barcoded library assemblies were transformed into TOP10 electrocompetent E. coli and a larger culture for plasmid purification was inoculated with ~6 × 104 and ~8 × 104 transformed cells for the libraries with lower and higher mutation frequency, respectively. These libraries were combined in a 1:12 ratio along with barcoded, catalytically inactivated TnpB protein controls before transformation into yeast.
Variant–barcode mapping
After library construction, variants were associated with their barcodes using long-read sequencing (PacBio Sequel II for the protein DMS library and Nanopore MinION for the reRNA DMS and combinatorial libraries). All reads were aligned to a reference plasmid and barcode sequences extracted using Minimap2 (version 2.26) and SAMtools (version 1.19)54,55. Subalignments were made for all reads with a given barcode and a consensus sequence was created using SAMtools for all barcodes with at least two reads for PacBio sequencing and at least ten reads for nanopore sequencing. Barcodes of incorrect length and consensus sequences containing nonprogrammed mutations were discarded. For the reRNA, protein and stacked libraries, 606, 7,766 and 6,592 variants were mapped, with an average of 33, 28 and 15 barcodes per variant, respectively. Full analysis scripts and processed data are available on GitHub (https://github.com/SavageLab/tnpb_dms).
Reporter yeast strain creation
Yeast ade2− reporter strains were created with the ‘delitto perfetto’ approach56. An intermediate ADE2 KO was derived from S. cerevisiae BY4741 (American Type Culture Collection (ATCC), 201388; Meyen ex E.C. Hansen) using the CORE cassette GSKU, excluding Gal-I-SceI. To create reporter strains, the intermediate strain was cotransformed with linearized DNA containing the target site flanked by duplicate homology regions and with plasmid carrying SpyCas9 targeting the CORE cassette. SpyCas9 was constitutively expressed, triggering DSBs in the CORE cassette and repair with the linear DNA template. Target site integration was confirmed by PCR amplification and Sanger sequencing and the strain was cured of the Cas9 plasmid. Using this approach, we generated ade2− reporter strains UniPAM1, UniPAM2 (target 1 strain) and UniPAM5 (target 2 strain).
Yeast pooled library selection assays
Plasmid DNA was linearized by PaqCI digestion before transformation and expression vectors were assembled by gap repair homologous recombination in yeast. Linearized plasmid libraries and backbone plasmid were transformed in a 1:3 molar ratio. For each experimental replicate, 4–5 µg of total linearized plasmid containing the DMS or combinatorial libraries of the TnpB protein was transformed. For the reRNA DMS library, 1.5 µg of total linearized plasmid library was transformed. Yeast were transformed with the lithium acetate and single-stranded carrier DNA/PEG method57.
After transformation, cells were resuspended in synthetic complete dropout medium (SCD) lacking leucine to select for transformation and gap repair of plasmids and recovered overnight at 30 °C. The following morning, a fraction of the culture was removed for a preinduction time point. The remaining cells were induced in liquid medium lacking leucine with 2% galactose(w/v) at an initial optical density at 600 nm (OD600) of 1.0. Induced cultures grew at 30 °C and culture samples were removed at multiple time points, pelleted, washed in milliQ water and plated on selective (−adenine −leucine) and nonselective (+adenine −leucine) SCD solid medium on bioassay dishes (Thermo Fisher). Several cell concentrations were plated at each time point to ensure maximal library coverage. Before plating, all cultures were grown with supplemental (160 mg ml−1) adenine. Yeast plates were incubated at 30 °C for 48 h, after which colonies were scraped and plasmid DNA was extracted using Zymoprep yeast plasmid miniprep II (Zymo Research). Barcodes were amplified from plasmid DNA using KAPA HiFi HotStart ReadyMix (Roche) with 6–12 cycles for PCR1 and 10 cycles for PCR2. PCRs were cleaned up with Ampure XP beads (Beckman Coulter) and submitted for 150-bp paired-end sequencing on Illumina NextSeq sequencer at the Innovative Genomics Institute (IGI) NGS sequencing core.
Variant enrichment calculations
Barcode enrichment was assessed by calculating the log ratio of reads containing a given barcode in selective and nonselective samples. Barcodes with fewer than five reads in selective or nonselective conditions were removed from analysis and the log ratio was normalized by the total number of reads in selective and nonselective sequencing samples. For the protein and stacked protein libraries, variant enrichment was calculated as the median barcode enrichment for all barcodes associated with a given variant. For the reRNA library, variant enrichment was calculated as the mean of all barcode enrichments as this produced higher replicate correlation. Variant enrichments were normalized to WT such that WT had an enrichment value of zero. A two-sided Mann–Whitney test was performed to calculate the statistical significance and effect size for each variant for each replicate. Variant enrichments are represented in plots generated with DataGraph (version 5.4) and Plotly (version 5.24.1).
Yeast cleavage assays
To compare the activities of TnpB variants and orthologs, as well as of CRISPR–Cas effectors, yeast cells were transformed with 0.5–1.5 µg of clonal plasmids or linearized DNA for assembly of clonal plasmids and induced as described above. At the preinduction and postinduction time points, approximately 1–3 OD600 units were removed from the transformed yeast culture, washed with milliQ water, resuspended in 200 µl of milliQ water and serially diluted in triplicate. Serial dilutions were plated on selective (−adenine −leucine) and nonselective (+adenine −leucine) solid SCD medium 8 h after induction, unless otherwise specified. Plates were incubated at 30 °C for 48 h, after which colony counts from serial dilutions were used to estimate the total number of CFUs. Colony reversion was calculated by dividing the number of CFUs on selective medium over the number of CFUs on nonselective medium and multiplied by 100 for the percentage. Colony reversion percentages were plotted as bar graphs with GraphPad Prism (version 10.6.1).
TnpB protein western blots in yeast
At 24 h after induction of a yeast cleavage assay, 2.5 OD600 units of yeast cells were harvested for western blots at 3,000g for 5 min. Cells were resuspended in 100 μl of milliQ water before lysis by adding 100 μl 0.2 M NaOH and incubation at room temperature for 5 min. Cell lysate was pelleted by centrifugation at 21,000g for 2 min, washed with 200 μl of 1× PBS and pelleted again at 21,000g for 2 min. Pellets were resuspended in 30 μl of 1× PBS and 30 μl of 4× Laemmli buffer (62.5 mM Tris-HCl, pH 6.8, 10% glycerol, 1% LDS and 0.005% bromophenol blue) (BioRad). Samples were boiled at 95 °C for 3 min before 12 μl of supernatant was loaded for SDS–PAGE on a 4–20% Criterion TGX precast Midi protein gel and separated at 125 V for 60 min. Transfer to a BioRad Trans-Blot Turbo Midi PVDF transfer pack was performed with the Trans-Turbo turbo transfer system. The membrane was blocked with 5% milk in Tris-buffered saline with Tween-20 (TBST) for 1 h at room temperature and then incubated overnight with mouse anti-FLAG (1:10,000; Sigma-Aldrich, F3165, lot SLCP4941) and rabbit anti-PGK1 (phosphoglycerate kinase 1; 1:30,000; provided by J. Thorner58) in 2.5% milk in TBST overnight at 4 °C. The membrane was washed three times with TBST for 10 min each at room temperature, then incubated with goat anti-mouse (1:10,000; LiCor, 926-32210, lot D40409-05) secondary antibody and imaged. The membrane was then washed three times and then incubated with goat anti-rabbit (1:30,000; LiCor, 926-68071, lot D40416-05) secondary antibody for 1 h at room temperature. The membrane was washed again as described before. Images were acquired on the LiCor Odyssey CLx and processed by using Image Studio version 6.1.
Comparative sequence and structure alignments
A multiple-sequence alignment (MSA) was created using an established EVcouplings (version 0.2.1) pipeline59 with HMMER (version 3.4). The ISDra2 TnpB protein sequence was used as the Jackhmmer query for five search iterations against the UniRef90 database with a domain and sequence bit score threshold of 0.1. Redundant sequences were removed using HHfilter and sequences with less than 50% coverage were removed. Per-position amino acid frequencies were determined by calculating the ratio of the amino acid’s occurrence to the total number of sequences with a residue present at that position10.
Structural alignments of ISDra2, ISYmu1 and ISAba30 TnpBs were generated with FoldMason60 (shown) and pairwise alignments were generated with TM-align61 with the AlphaFold2-predicted WT amino acid sequences62. Similarity was calculated with a Blosum62 matrix with a threshold of 1 (ref. 24).
Mammalian genome editing
Mammalian cell culture experiments were performed in the HEK293T cell line (ATCC, CRL-3216; supplied by the University of California (UC) Berkeley Cell Culture Facility) and HEK293T EGFP (a gift from K. Chen, UC Berkeley)17. Mammalian cells (HEK393T or HEK293T-GFP) were grown in DMEM with high glucose, GlutaMAX supplement and pyruvate (Thermo Fisher) supplemented with 10% FBS (Avantor Seradigm) at 37 °C and 5% CO2.
Cells were seeded at approximately 10,000 cells per well in 96-well plates 16–24 h before transfection. The transfection mix was prepared by combining plasmids encoding the protein and reRNA/sgRNA (100 ng carrying TnpB and 145 ng carrying SpyCas9 for 2.6 fmol per transfection) with 9 µl Opti-MEM I reduced-serum medium (Thermo Fisher) and 0.3 µl of TransIT-293 per transfection. Transfection mixes were incubated at room temperature for 30 min and added dropwise to the cells.
For flow cytometry, transfected plates were passaged 2 days after transfection and then harvested for flow cytometry after 2–5 days. Cells were trypsinized with 30 µl of 0.25% trypsin + EDTA was added to cells for 5 min at 37 °C and quenched with 120 µl of 1× PBS. Cells were transferred to 96-well round-bottom plates and analyzed by flow cytometry on an Attune NxT Flow Cytometer with an autosampler. Data were collected with Attune Cytometric Software (version 5.1.1) and analyzed using FlowJo software version 10.10.0 (Supplementary Data Fig. 2b).
For sequencing, cells were harvested 4 days after transfection and lysed with QuickExtract (Lucigen) according to the manufacturer’s instructions. Lysate was used directly for PCR. PCR products were cleaned with Ampure XP beads (Beckman Coulter), analyzed by a 4150 TapeStation (Agilent) and submitted for 150-bp or 300-bp paired-end sequencing on MiSeq or NextSeq sequencer at the IGI NGS sequencing core. The frequencies of the mutations were assessed by CRISPResso2 (version 2.3.1)63. Editing data were plotted as bar graphs with GraphPad Prism (version 10.6.1).
Off-target analysis
To assess the specificity of TnpB and TnpB variants, CRISPR RGEN Tools (Cas-OFFinder, version 2.4.1; http://www.rgenome.net/cas-offinder/) was used to predict potential genomic off-target sites containing the ‘TTGAT’ TAM/PAM and 2–6 mismatches in the target sequence27. Primers were designed using NCBI Primer-BLAST (https://www.ncbi.nlm.nih.gov/tools/primer-blast/).
N. benthamiana editing
All plasmid vectors were delivered to N. benthamiana by Agrobacterium tumefaciens strain GV3101 infiltration. Cultures containing the vector of interest were grown in lysogeny broth (LB) medium supplemented with spectinomycin (50 µg ml−1), gentamicin (30 µg ml−1) and rifampicin (25 µg ml−1) overnight at 30 °C. The next day, cultures were spun down at 3,500g for 10 min. The pellet was then resuspended in infiltration media (10 mM MgCl2, 10 mM MES pH 5.6 and 150 µM acetosyringone, in milliQ water) and diluted to an OD600 of 1.0. The resuspension was incubated at room temperature for 3 h before infiltration.
Syringe infiltration was performed on the abaxial surface of leaves of 4-week-old N. benthamiana plants. Infiltrated plants were then watered and transferred back to the plant growth chamber (Percival) (16-h light, 8-h dark photoperiod, 80 µmol m−2 light intensity, 50% humidity, at 23 °C) for 4 days. After the 4-day period, leaf discs were taken for each infiltrated leaf using a hole puncher tool (Electron Microscopy Sciences, 6903950). The obtained leaf tissue was lysed in 700 µl of 2% CTAB (10 g CTAB, 100 mM Tris-HCl, 20 mM EDTA, 1.4 M NaCl, 1% polyvinylpyrrolidone) or 20 µl of Phire plant direct PCR dilution buffer (Thermo Scientific, F160S), following flash-freezing in liquid nitrogen. Leaf lysate was used directly for PCR reactions or used for genomic DNA extraction64.
PCR reactions were performed using Phire plant direct PCR master mix (Thermo Fischer) and PCR products were cleaned with Ampure XP beads (Beckman Coulter), analyzed by a 4150 TapeStation (Agilent) and submitted for 150-bp or 300-bp paired-end sequencing on MiSeq or NextSeq sequencer at the IGI NGS sequencing core. The frequencies of the mutations were assessed by CRISPResso (version 2.3.1)63.
Rice editing
Transgenic callus tissues and plants were generated by Agrobacterium-mediated transformation using established protocols65 with minor modifications. Mature seeds of rice (O. sativa L. japonica cv. Kitaake) were dehulled and surface-sterilized for 3 min with 70% (v/v) ethanol followed by 15 min in 20% (v/v) commercial bleach (5.25% sodium hypochlorite v/v) containing one drop of Tween-20. Seeds were washed three times with sterile water to remove residual bleach. Sterilized seeds were placed on callus induction medium (CIM)65 without BAP and incubated in the dark at 28 °C to initiate callus induction. High-quality calli were selected and transferred to fresh CIM for proliferation.
A total of 50 pieces of 6–8-week-old calli, approximately 2–3 mm in diameter, were dried on empty sterile Petri dishes for 30 min before incubation with an A. tumefaciens AGL1 suspension (OD600nm = 0.2) carrying each transformation vector. TnpB and reRNA were cloned into the pKb-TnpB2 vector used by Karmakar et al. to deliver WT ISDra2 TnpB to rice callus38. After a 30-min incubation, the Agrobacterium suspension was removed. Calli were then placed on sterile filter paper, transferred to cocultivation medium65 and incubated in the dark at 21 °C for 3 days. Calli were then transferred to resting medium66 (OsCIM2 supplemented with 150 mg l−1 cefotaxime and 100 mg l−1 timentin) and incubated in the dark at 28 °C for 7 days. Calli were then transferred to the selection medium (resting medium plus 40 mg l−1 hygromycin B) and incubated in the dark at 28 °C. Tissues were transferred to the fresh selection medium every 2 weeks. The remaining callus tissues were moved to regeneration media65 containing 40 mg l−1 hygromycin B and incubated at 26 °C, under a 16-h light (90 μmol of photon per m2 per s), 8-h dark photoperiod at 26 °C. When regenerated plantlets reached approximately 1 cm in height, they were transferred to 100 ml of rooting medium65 containing 20 mg l−1 hygromycin B and incubated at 26 °C under conditions of 16-h light (100–150 μmol of photons per m2 per s), 8-h dark photoperiod until roots were established and leaves touched the Phytatray II lid (Sigma-Aldrich) and leaf tissue was sampled from at least four leaves from each independent transformation event for sequencing. Collected leaf samples were crushed in 50 μl of dilution buffer included in the Phire plant direct PCR kit (Thermo Scientific), then centrifuged at 18,000g in an Eppendorf centrifuge for 5 min, stored at −80 °C and used directly for genotyping.
Leaf extract was used directly for PCR with Phire plant direct PCR master mix (Thermo Scientific) according to the manufacturer’s instructions. PCR products were cleaned with Ampure XP beads (Beckman Coulter), analyzed by a 4150 TapeStation (Agilent) and submitted for 300 bp paired-end sequencing on MiSeq or NextSeq sequencer at the IGI NGS sequencing core. The frequencies of the mutations were assessed by CRISPResso2 (ref. 63) and indel frequencies were averaged for leaf tissue samples originating from the same transformation event.
Pepper editing
Serrano Tampiqueno pepper (C. annuum L.) was grown in a growth chamber set at 24 °C with a 12-h light, 12-h dark cycle, a light intensity of 100 μE per m2 per s and 50% humidity. The Agrobacterium GV3101 strain, carrying various TnpB and reRNA with target sequences, was grown in LB medium supplemented with SRG (spectinomycin at 50 μg ml−1, rifampicin at 25 μg ml−1 and gentamicin at 50 μg ml−1) at 28 °C for 14–16 h. Agrobacterium cells were pelleted by centrifugation at 3,500g for 15 min, resuspended in infiltration medium containing 10 mM MgCl2, 10 mM MES and 250 μM 3′,5′-dimethoxy-4′-hydroxyacetophenone (acetosyringone) in milliQ water to an OD600 of 1.0 and incubated with gentle shaking for at least 3 h at room temperature. Agrobacterium was infiltrated into two fully expanded cotyledons of 20-day-old pepper seedlings using a 1-ml needleless syringe. For CaCHLHt4 and CaBRI1t11 infiltration, Agrobacterium with RNA silencing suppressor p19 was added at an OD600 of 0.05. Infiltrated plants were kept on growth light carts and, 4 days later, leaf samples were collected and crushed in 35 μl of dilution buffer included in the Phire plant direct PCR kit (Thermo Scientific), then centrifuged at 18,000g in an Eppendorf centrifuge for 5 min and stored at −80 °C or used directly for genotyping.
Next, 2 µl of leaf extract was used for PCR with Phire plant direct PCR master mix (Thermo Scientific) according to the manufacturer’s instructions. PCR products were cleaned with Ampure XP beads (Beckman Coulter), analyzed by a 4150 TapeStation (Agilent) and submitted for 300 bp paired-end sequencing on MiSeq or NextSeq sequencer at the IGI NGS sequencing core. The frequencies of the mutations were assessed by CRISPResso2 (ref. 63).
Nucleic acid and plasmid preparation
All DNA oligonucleotides used in this study, unless otherwise indicated, were synthesized by Integrated DNA Technologies. Plasmids, unless otherwise indicated, were assembled by Golden Gate cloning.
Reporting summary
Further information on research design is available in the Nature Portfolio Reporting Summary linked to this article.
Data availability
Raw sequencing data are available under BioProject PRJNA1422494. All other data (primers, RNA, protein sequences, variant enrichments and editing efficiencies) are available from the Supplementary Information. Sequences for MSAs were taken from UniRef90. Additional relevant materials (such as plasmids) are available from the corresponding author upon reasonable request or from Addgene. Source data are provided with this paper.
Code availability
All code for this paper is available from GitHub (https://github.com/SavageLab/tnpb_dms).
References
-
Altae-Tran, H. et al. Diversity, evolution, and classification of the RNA-guided nucleases TnpB and Cas12. Proc. Natl Acad. Sci. USA 120, e2308224120 (2023).
-
Karvelis, T. et al. Transposon-associated TnpB is a programmable RNA-guided DNA endonuclease. Nature 599, 692–696 (2021).
-
Altae-Tran, H. et al. The widespread IS200/IS605 transposon family encodes diverse programmable RNA-guided endonucleases. Science 374, 57–65 (2021).
-
Shmakov, S. et al. Diversity and evolution of class 2 CRISPR–Cas systems. Nat. Rev. Microbiol. 15, 169–182 (2017).
-
Makarova, K. S. et al. Evolutionary classification of CRISPR–Cas systems: a burst of class 2 and derived variants. Nat. Rev. Microbiol. 18, 67–83 (2020).
-
Sasnauskas, G. et al. TnpB structure reveals minimal functional core of Cas12 nuclease family. Nature 616, 384–389 (2023).
-
Nakagawa, R. et al. Cryo-EM structure of the transposon-associated TnpB enzyme. Nature 616, 390–397 (2023).
-
Koonin, E. V., Gootenberg, J. S. & Abudayyeh, O. O. Discovery of diverse CRISPR–Cas systems and expansion of the genome engineering toolbox. Biochemistry 62, 3465–3487 (2023).
-
Fowler, D. M. & Fields, S. Deep mutational scanning: a new style of protein science. Nat. Methods 11, 801–807 (2014).
-
Prywes, N. et al. A map of the rubisco biochemical landscape. Nature 638, 828–828 (2025).
-
Stiffler, M. A., Hekstra, D. R. & Ranganathan, R. Evolvability as a function of purifying selection in TEM-1 β-lactamase. Cell 160, 882–892 (2015).
-
Meers, C. et al. Transposon-encoded nucleases use guide RNAs to promote their selfish spread. Nature 622, 863–871 (2023).
-
Casini, A. et al. A highly specific SpCas9 variant is identified by in vivo screening in yeast. Nat. Biotechnol. 36, 265–271 (2018).
-
Li, Z. et al. Engineering a transposon-associated TnpB–ωRNA system for efficient gene editing and phenotypic correction of a tyrosinaemia mouse model. Nat. Commun. 15, 831 (2024).
-
Antao, V. P., Lai, S. Y. & Tinoco, I. Jr. A thermodynamic study of unusually stable RNA and DNA hairpins. Nucleic Acids Res. 19, 5901–5905 (1991).
-
Banáš, P. et al. Performance of molecular mechanics force fields for RNA simulations: Stability of UUCG and GNRA hairpins. J. Chem. Theory Comput. 6, 3836–3849 (2010).
-
Chen, K. et al. Lung and liver editing by lipid nanoparticle delivery of a stable CRISPR–Cas9 ribonucleoprotein. Nat. Biotechnol. 43, 1445–1457 (2025).
-
Kleinstiver, B. P. et al. Engineered CRISPR–Cas12a variants with increased activities and improved targeting ranges for gene, epigenetic and base editing. Nat. Biotechnol. 37, 276–282 (2019).
-
Zhang, H. et al. An engineered xCas12i with high activity, high specificity, and broad PAM range. Protein Cell 14, 538–543 (2023).
-
Chen, Y. et al. Synergistic engineering of CRISPR–Cas nucleases enables robust mammalian genome editing. Innovation (Camb.) 3, 100264 (2022).
-
Hino, T. et al. An AsCas12f-based compact genome-editing tool derived by deep mutational scanning and structural analysis. Cell 186, 4920–4935 (2023).
-
Xiang, G. et al. Evolutionary mining and functional characterization of TnpB nucleases identify efficient miniature genome editors. Nat. Biotechnol. 42, 745–757 (2024).
-
Zhang, Y. & Skolnick, J. Scoring function for automated assessment of protein structure template quality. Proteins 57, 702–710 (2004).
-
Henikoff, S. & Henikoff, J. G. Amino acid substitution matrices from protein blocks. Proc. Natl Acad. Sci. USA 89, 10915–10919 (1992).
-
Wrenbeck, E. E. et al. Plasmid-based one-pot saturation mutagenesis. Nat. Methods 13, 928–930 (2016).
-
Mighell, T. L., Toledano, I. & Lehner, B. SUNi mutagenesis: scalable and uniform nicking for efficient generation of variant libraries. PLoS ONE 18, e0288158 (2023).
-
Bae, S., Park, J. & Kim, J.-S. Cas-OFFinder: a fast and versatile algorithm that searches for potential off-target sites of Cas9 RNA-guided endonucleases. Bioinformatics 30, 1473–1475 (2014).
-
Marquart, K. F. et al. Effective genome editing with an enhanced ISDra2 TnpB system and deep learning-predicted ωRNAs. Nat. Methods 21, 2084–2093 (2024).
-
Tokuriki, N. & Tawfik, D. S. Stability effects of mutations and protein evolvability. Curr. Opin. Struct. Biol. 19, 596–604 (2009).
-
Pixley, K. V. et al. Genome-edited crops for improved food security of smallholder farmers. Nat. Genet. 54, 364–367 (2022).
-
Nasti, R. A. & Voytas, D. F. Attaining the promise of plant gene editing at scale. Proc. Natl Acad. Sci. USA 118, e2004846117 (2021).
-
Ellison, E. E. et al. Multiplexed heritable gene editing using RNA viruses and mobile single guide RNAs. Nat. Plants 6, 620–624 (2020).
-
Ali, Z. et al. Efficient virus-mediated genome editing in plants using the CRISPR/Cas9 system. Mol. Plant 8, 1288–1291 (2015).
-
Lv, Z. et al. Targeted mutagenesis in Arabidopsis and medicinal plants using transposon-associated TnpB. J. Integr. Plant Biol. 66, 2083–2086 (2024).
-
Zhang, R. et al. IsDge10 is a hypercompact TnpB nuclease that confers efficient genome editing in rice. Plant Commun. 5, 101068 (2024).
-
Weiss, T. et al. Viral delivery of an RNA-guided genome editor for transgene-free germline editing in Arabidopsis. Nat. Plants 11, 967–976 (2025).
-
Kannan, S. et al. Evolution-guided protein design of IscB for persistent epigenome editing in vivo. Nat. Biotechnol. https://doi.org/10.1038/s41587-025-02655-3 (2025).
-
Karmakar, S. et al. A miniature alternative to Cas9 and Cas12: transposon-associated TnpB mediates targeted genome editing in plants. Plant Biotechnol. J. 22, 2950–2953 (2024).
-
Li, Q. et al. Genome editing in plants using the TnpB transposase system. aBIOTECH 5, 225–230 (2024).
-
Kumar, M. et al. Breaking the glass ceiling of stable genetic transformation and gene editing in the popular pepper cv Cayenne. J. Exp. Bot. 76, 2688–2699 (2025).
-
Hickman, A. B. et al. DNA recognition and the precleavage state during single-stranded DNA transposition in D. radiodurans. EMBO J. 29, 3840–3852 (2010).
-
Chandler, M. et al. Breaking and joining single-stranded DNA: the HUH endonuclease superfamily. Nat. Rev. Microbiol. 11, 525–538 (2013).
-
Yoon, P. H. et al. Eukaryotic RNA-guided endonucleases evolved from a unique clade of bacterial enzymes. Nucleic Acids Res. 51, 12414–12427 (2023).
-
Touchon, M. & Rocha, E. P. C. Causes of insertion sequences abundance in prokaryotic genomes. Mol. Biol. Evol. 24, 969–981 (2007).
-
Doolittle, W. F., Kirkwood, T. B. & Dempster, M. A. Selfish DNAs with self-restraint. Nature 307, 501–502 (1984).
-
Cheng, P. et al. Zero-shot prediction of mutation effects with multimodal deep representation learning guides protein engineering. Cell Res. 34, 630–647 (2024).
-
Bershtein, S. & Tawfik, D. S. Ohno’s model revisited: measuring the frequency of potentially adaptive mutations under various mutational drifts. Mol. Biol. Evol. 25, 2311–2318 (2008).
-
Tokuriki, N., Stricher, F., Schymkowitz, J., Serrano, L. & Tawfik, D. S. The stability effects of protein mutations appear to be universally distributed. J. Mol. Biol. 369, 1318–1332 (2007).
-
Notin, P. et al. ProteinGym: large-scale benchmarks for protein fitness prediction and design. In Proc. 37th International Conference on Neural Information Processing Systems (eds Oh, A. et al.) (ACM, 2023).
-
Wiegand, T. et al. TnpB homologues exapted from transposons are RNA-guided transcription factors. Nature 631, 439–448 (2024).
-
Wang, J. Y. & Doudna, J. A. CRISPR technology: a decade of genome editing is only the beginning. Science 379, eadd8643 (2023).
-
Xu, X. et al. Engineered miniature CRISPR–Cas system for mammalian genome regulation and editing. Mol. Cell 81, 4333–4345.e4 (2021).
-
Nagalakshmi, U. et al. High-efficiency, transgene-free plant genome editing by viral delivery of an engineered TnpB. Nat. Plants https://doi.org/10.1038/s41477-026-02237-4 (2026).
-
Li, H. Minimap2: pairwise alignment for nucleotide sequences. Bioinformatics 34, 3094–3100 (2018).
-
Li, H. et al. The Sequence Alignment/Map format and SAMtools. Bioinformatics 25, 2078–2079 (2009).
-
Stuckey, S. & Storici, F. Gene knockouts, in vivo site-directed mutagenesis and other modifications using the delitto perfetto system in Saccharomyces cerevisiae. Methods Enzymol. 533, 103–131 (2013).
-
Gietz, R. D. & Schiestl, R. H. High-efficiency yeast transformation using the LiAc/SS carrier DNA/PEG method. Nat. Protoc. 2, 31–34 (2007).
-
Baum, P., Thorner, J. & Honig, L. Identification of tubulin from the yeast Saccharomyces cerevisiae. Proc. Natl Acad. Sci. USA 75, 4962–4966 (1978).
-
Hopf, T. A. et al. The EVcouplings Python framework for coevolutionary sequence analysis. Bioinformatics 35, 1582–1584 (2019).
-
Gilchrist, C. L. M., Mirdita, M. & Steinegger, M. Multiple protein structure alignment at scale with FoldMason. Science 391, 485–488 (2026).
-
Bittrich, S., Segura, J., Duarte, J. M., Burley, S. K. & Rose, Y. RCSB protein Data Bank: exploring protein 3D similarities via comprehensive structural alignments. Bioinformatics 40, btae370 (2024).
-
Jumper, J. et al. Highly accurate protein structure prediction with AlphaFold. Nature 596, 583–589 (2021).
-
Clement, K. et al. CRISPResso2 provides accurate and rapid genome editing sequence analysis. Nat. Biotechnol. 37, 224–226 (2019).
-
Yu, D. et al. An easily-performed high-throughput method for plant genomic DNA extraction. Anal. Biochem. 569, 28–30 (2019).
-
Karavolias, N. G. et al. Paralog editing tunes rice stomatal density to maintain photosynthesis and improve drought tolerance. Plant Physiol. 192, 1168–1182 (2023).
-
Poddar, S., Tanaka, J., Cate, J. H. D., Staskawicz, B. & Cho, M.-J. Efficient isolation of protoplasts from rice calli with pause points and its application in transient gene expression and genome editing assays. Plant Methods 16, 151 (2020).
-
He, S. et al. The IS200/IS605 family and ‘peel and paste’ single-strand transposition mechanism. Microbiol. Spectr. https://doi.org/10.1128/microbiolspec.mdna3-0039-2014 (2015).
Acknowledgements
We thank J. L. Rivera for cultivating the N. benthamiana plants used in this study, and thank N. Prywes for advice on library design and general guidance on DMS and data analysis. We thank L. Oltrogge for key assistance with data analysis and sequencing, H. Chang for assistance with obtaining rice tissue samples, and K. Chen for providing the HEK293T EGFP cell line used for EGFP KO assays. We thank J. Desmarais, B. Cress, A. Eggers and J. Cofsky for establishing protocols and providing components of expression vectors that we used in the yeast cleavage assay. Antiserum against PGK was provided by J. Thorner58. We thank N. Krishnappa for assistance with running NGS samples, and J. Turnšek for help with preliminary protein purification. We also thank O. Tuck for insights into the protein DMS dataset and experimental design, and M. Lukarska for reviewing the manuscript. This material is based upon work supported by the National Science Foundation graduate research fellowship program under grant no. DGE 2146752 (B.W.T., R.F.W. and C.I.T.). Any opinions, findings and conclusions or recommendations expressed in this material are those of the authors and do not necessarily reflect the views of the National Science Foundation. R.V.T. was funded by the Rose Hills Foundation as part of UC Berkeley’s summer undergraduate research fellowship program. B.T.D. was funded by UC Berkeley’s Haas Scholars Program. J.P. is a Howard Hughes Medical Institute (HHMI) fellow of The Jane Coffin Childs Memorial Fund. D.F.S. is an investigator of the HHMI and this research was funded by National Institutes of Health grant no. 1R35GM158173. J.A.D. is an investigator of the HHMI and this research was supported by the HHMI. Gene-editing research in the S.P.D.-K. lab is supported by the National Science Foundation grant no. IOS-2303522 and the IGI.
Ethics declarations
Competing interests
D.F.S. is a cofounder and scientific advisory board member of Scribe Therapeutics. The Regents of the UC have patents issued and pending for CRISPR technologies on which J.A.D. is an inventor. J.A.D. is a cofounder of Azalea Therapeutics, Caribou Biosciences, Editas Medicine, Evercrisp, Scribe Therapeutics and Mammoth Biosciences. J.A.D. is a scientific advisory board member at Isomorphic Labs, BEVC Management, Evercrisp, Caribou Biosciences, Scribe Therapeutics, Mammoth Biosciences, The Column Group and Inari. J.A.D. is also an advisor for Aditum Bio. J.A.D. is chief science advisor to Sixth Street and a director at Johnson & Johnson, Altos and Tempus. B.W.T., R.F.W., C.I.T., J.E.R., U.N., S.P.D.-K., D.F.S. and J.A.D. have submitted related patents. R.V.T. is currently an employee of Scribe Therapeutics. The remaining authors declare no competing interests.
Peer review
Peer review information
Nature Biotechnology thanks Hiroshi Nishimasu and the other, anonymous, reviewer(s) for their contribution to the peer review of this work.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Extended data
Extended Data Fig. 1 In vivo yeast cleavage assay captures a range of RNA-guided endonuclease activity across TnpB, Cas9, and Cas12 endonucleases.
a, Assessment of Cas9, Cas12, and ISDra2 TnpB RNA-guided endonuclease activity in yeast cleavage assays, with (+) or without (-) the gRNA or reRNA. ISDra2 TnpB protein was expressed from either the non-codon-optimized open reading frame from D. radiodurans R1 (GenBank AE000513.1), or TnpB was codon optimized for high frequency codon usage between H. sapiens and S. cerevisiae. Data are plotted as the mean and s.e.m. (standard error of mean) from technical triplicate titer plating measurements (n = 3). b, Schematic representation of the ISDra2 Trim2 reRNA variant (red rArA bases replacing ΔrC−50-rU−69). Color scheme corresponds to Fig. 2a. c, ISDra2 TnpB endonuclease activity in yeast with various reRNA scaffold lengths, including 231 nts, 116 nts, and the reported Trim2 reRNA variant. TnpB and reRNA were expressed in a yeast strain with an reRNA-complementary target site (on), or in a yeast strain with a non-complementary target site (non). Data are plotted as the mean and s.e.m. from technical triplicate titer plating measurements (n = 3).
Extended Data Fig. 2 Distribution of enriched amino acid substitutions varies by TnpB domain.
a, (Left) Histogram and box plots of the enrichment values for all protein DMS library mutations, grouped by ISDra2 TnpB domain. Box plots show the median (center line), interquartile range (box), and whiskers extending to Q1 − 1.5×IQR and Q3 + 1.5×IQR. Outliers (Q1 − 1.5×IQR or Q3 + 1.5×IQR) are drawn as circles and extreme outliers (Q1 − 3×IQR or Q3 + 3×IQR) are drawn as open circles. Summary statistics for each group are provided in Supplementary Data 3. (Right) ISDra2 domains (top right) and max enrichment per amino acid residue (bottom right) mapped onto the surface of ISDra2 TnpB ternary structure (PDB ID: 8EXA). b, Enrichment scores averaged for all stop codon mutations plotted across the length of the protein. Enrichment scores are not normalized to WT. The dashed line indicates position 376, marking the C-terminus of the minimal active TnpB truncation variant (Δ376–408) previously identified7.
Extended Data Fig. 3 Positively charged amino acids are enriched near nucleic acid contacts.
a, Enrichment of positively (R, K) and negatively (D, E) charged amino acid substitutions for residues proximal to nucleic acid. To assess the impact of amino acid substitutions near nucleic acids, we defined proximal residues as those with Cα atoms within 8 Å from nucleic acid atoms. This cutoff ensured inclusion of previously identified direct interactions and potential contacts by R/K/D/E mutations. Box plots show the median (center line), interquartile range (box), and whiskers extending to Q1 − 1.5×IQR and Q3 + 1.5×IQR. Summary statistics for each group are provided in Supplementary Data 3. b, The average enrichment for substitutions to positively charged (R, K) or negatively charged (D, E) amino acids was calculated at each position. If the WT residue was already R/K/D/E, its enrichment value was included in the average as zero. Enrichment values were mapped onto the ISDra2 TnpB cryo-EM ternary structure6, with additional surface coloring by domain to help orient the reader to the structural context.
Extended Data Fig. 4 Activating mutations found for ISDra2 TnpB are transferable to TnpB orthologs.
a, Multiple structural alignment of ISDra2, ISYmu1, and ISAba30 TnpB. b, c, Activity of ortholog mutants was assessed by percent colony reversion with the yeast cleavage assay and compared to WT orthologs and negative non-complementary reRNA-target controls. b, Data represent mean ± s.e.m. (n = 3 technical plating replicates 8 h post-induction). c, Titer plates (b) where each of the protein variants have been expressed in S. cerevisiae for 8 h and titer plated on selective (-adenine) and nonselective (+adenine) media. Technical pipetting replicates from titer plates are shown.
Extended Data Fig. 5 Combinatorial library construction, experimental enrichment, and distribution of mutation number.
a, Schematic of library construction using pooled nicking mutagenesis. Plasmid was digested for ssDNA template, and 33 selected amino acid mutations at 19 positions were introduced on ssDNA oligos as described in Methods. Two libraries with low and high mutation frequencies were combined, with an average of 3 and 5 mutations, respectively. b, Volcano plots of variant enrichment and statistical significance in orthogonal reporter yeast strains with different target sites. Enrichments are shown from 4 h and 8 h post-induction. Enrichment is calculated by averaging two biological replicates. Significance was calculated from individual barcode enrichments per variant relative to wild-type (two-sided Mann-Whitney U-test). c, Enrichment score distributions separated by variant mutation number for experimental conditions in b.
Extended Data Fig. 6 Western blots showing that expression levels of enhanced TnpB variants do not increase in S. cerevisiae.
a, (Top) Construct design for expression of ISDra2 TnpB WT protein and variants with an NLS and FLAG tag in yeast. (Bottom) Western blot from yeast lysate with anti-FLAG antibody and with an anti-PGK1 antibody as a loading control. Western blot was repeated for WT ISDra2, eTnpBa, eTnpBc, and eTnpBe with similar results. b, Activity of each variant was assessed by colony reversion in the yeast cleavage assay. Data represent mean ± s.e.m. (n = 3 technical plating replicates).
Extended Data Fig. 7 Assessment of combinatorial TnpB variant off and on-target editing, with reRNA mutants in HEK293Ts.
a, Indel frequency of six combinatorial variants at genomic loci in HEK293T cells, with WT ISDra2 TnpB, WT ISYmu1 TnpB, and no plasmid (NC) controls. Indel frequencies for eTnpBa-eTnpBe and WT TnpB at TET1, PGK1, AGBL1, and VEGFA are also represented in Fig. 5b. Data are plotted as the mean and s.e.m. from biological replicates (n = 3). ND indicates no data. Stars indicate a statistically significant increase in indel frequencies compared to WT ISDra2 TnpB as calculated using a two-sided unpaired Student’s t-test. (Significance: *, **, *** for p ≤ 0.05, 0.01, 0.001, respectively). b, Indel frequency of WT and combinatorial variant TnpBs at off-target sites identified by Cas-OFFinder, with 4–6 mismatches to three on-target sites. Sample order and color scheme match a. Off-target sequences (non-target strand) are listed, with TAM in blue and reRNA-target mismatches in red. Data are plotted as the mean and s.e.m. from biological replicates (n = 3). c, Pairs of reRNA deletion and ISDra2 TnpB protein mutants were tested with the EGFP KO assay, where EGFP-negative cells were measured by flow cytometry seven days after transfection. Data are presented as the mean ± s.e.m. from biological replicates (n = 3).
Extended Data Fig. 8 TnpB protein and reRNA mutants enable increases in TnpB-mediated indel frequencies in N. benthamiana.
a, Indel frequencies of TnpB combinatorial protein mutants at NbPDS1-2. Data represent the mean of n = 2 independent agroinfiltrations (biological replicates), except for eTnpBb and eTnpBd, where n = 3. b, Indel frequencies of TnpB reRNA and protein mutants at PDS1-1 and PDS1-2 sites in N. benthamiana. Data represent the mean of n = 2 independent agroinfiltrations. NC, negative control.
Extended Data Fig. 9 eTnpBc and eTnpbe are specific, highly active RNA-guided endonucleases in N. benthamiana and also show activity in pepper.
a, Indel frequencies of eTnpBc and eTnpBe compared to wild-type ISDra2 TnpB and a negative control (untransformed Agrobacterium infiltration) at Cas-OFFinder-predicted off-target sites in N. benthamiana. b, Indel frequencies of ISDra2 TnpB variants compared to wild-type ISYmu1TnpB, AsCas12f-HKRA, NovaIscB, and SpyCas9. Data are plotted as the mean and s.e.m. from biological replicates (n = 3 for all conditions, except the NbEDS1 Off3 ISDra2 where n = 2) in a, b. c, Indel frequencies at three genomic sites in pepper. Data are plotted as the mean and s.e.m. from biological replicates (n = 5 for all conditions except eTnpBe at CaPDS1, where n = 6). Stars indicate a statistically significant increase in indel frequencies compared to WT ISDra2 TnpB as calculated using a two-sided unpaired Student’s t-test. (Significance: *, **, *** for p ≤ 0.05, 0.01, 0.001, respectively) in b, c.
Extended Data Fig. 10 Deep mutational scanning of reRNA reveals mutational tolerance within the reRNA stem 1-RE overlap.
a, Box and whisker plots showing the distribution of log2 enrichment for single nucleotide substitutions, where nucleotide substitutions were grouped by reRNA region. Box plots show the median (center line), interquartile range (box), and whiskers extending to Q1 − 1.5×IQR and Q3 + 1.5×IQR. b, Log2 enrichment values for single-nucleotide substitutions grouped by reRNA position. Points denote individual mutants, the blue line indicates the mean, and vertical gray bars represent standard deviation where n ≥ 3. Summary statistics for each group are provided in Supplementary Data 3 for a, b. The x-axis includes annotations for both the overlapping RE DNA and reRNA sequences. Within the RE, key functional elements are highlighted, including the ssDNA subterminal hairpins recognized by TnpA for transposon excision, as well as the tetranucleotide cleavage (CR) and guide (GR) sequences, which form base-pairing interactions and direct TnpA cleavage42,67. Additionally, positions within the subterminal hairpin important for TnpA binding and strand discrimination are indicated in blue41.
Supplementary information
Source data
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Thornton, B.W., Weissman, R.F., Rodriguez, J.E. et al. Engineered TnpB genome editors for plants and human cells identified by ribonucleoprotein mutational scanning. Nat Biotechnol (2026). https://doi.org/10.1038/s41587-026-03059-7
-
Received:
-
Accepted:
-
Published:
-
Version of record:
-
DOI: https://doi.org/10.1038/s41587-026-03059-7